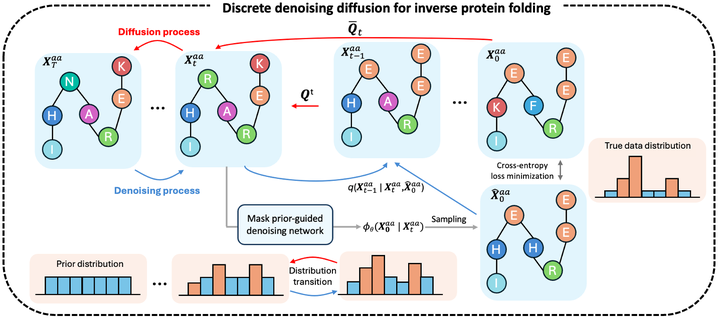
Inverse protein folding generates valid amino acid sequences that can fold into a desired protein structure, with recent deep-learning advances showing significant potential and competitive performance. However, challenges remain in predicting highly uncertain regions, such as those with loops and disorders. To tackle such low-confidence residue prediction, we propose a Mask prior-guided denoising Diffusion (MapDiff) framework that accurately captures both structural and residue interactions for inverse protein folding. MapDiff is a discrete diffusion probabilistic model that iteratively generates amino acid sequences with reduced noise, conditioned on a given protein backbone. To incorporate structural and residue interactions, we develop a graph-based denoising network with a mask prior pre-training strategy. Moreover, in the generative process, we combine the denoising diffusion implicit model with Monte-Carlo dropout to improve uncertainty estimation. Evaluation on four challenging sequence design benchmarks shows that MapDiff significantly outperforms state-of-the-art methods. Furthermore, the in-silico sequences generated by MapDiff closely resemble the physico-chemical and structural characteristics of native proteins across different protein families and architectures.
Peizhen Bai
PhD Student (now a Senior Machine Learning Scientist at AstraZeneca)
Filip Miljković
Associate Principal AI Scientist at AstraZeneca
Xianyuan Liu
Assistant Head of AI Research Engineering & Senior AI Research Engineer
Haiping Lu
Director of the UK Open Multimodal AI Network, Professor of Machine Learning, and Head of AI Research Engineering
I am a Professor of Machine Learning. I develop translational multimodal AI technologies for advancing healthcare and scientific discovery.